
社内資料(承認時評価資料):
日本人を含む国際共同第Ⅲ相臨床試験
Balwani M, Sardh E, Ventura P, et al.
N Engl J Med. 2020;382(24):2289-2301. (本試験はAlnylam Pharmaceuticalsの支援により実施された)
Ventura P, Bonkovsky HL, Gouya L, et al.
Liver Int. 2022;42(1):161-172. (本試験はAlnylam Pharmaceuticalsの支援により実施された)
Kuter DJ, Bonkovsky HL, Monroy S, et al.
J Hepatol. 2023;79(5):1150-1158. (本試験はAlnylam Pharmaceuticalsの支援により実施された)
目的
急性肝性ポルフィリン症(AHP)患者を対象にギブラーリの有効性及び安全性を評価する。
対象
AHP患者94例(日本人3例を含む)
- AHP[急性間欠性ポルフィリン症(AIP)、遺伝性コプロポルフィリン症(HCP)、異型ポルフィリン症(VP)又はALA脱水酵素欠損ポルフィリン症(ADP)]と診断された成人(18歳以上)及び青年期(12歳以上18歳未満)の患者
- 診断の根拠は、次の3つを満たすこととした。
- ①臨床的特徴を有する。
- ②スクリーニング前1年間又はスクリーニング期間中に、尿中又は血漿中のアミノレブリン酸(ALA)値又はポルフォビリノーゲン(PBG)値が基準範囲上限(ULN)の4倍以上であった記録を有する。
- ③ポルフィリン症関連遺伝子の変異の記録を有する。
ただし、ポルフィリン症関連遺伝子の変異が同定されていない場合、臨床的特徴が認められ、生化学的診断基準を満たしていれば本試験への参加に適格とした。
- スクリーニング前6ヵ月間に、入院、緊急受診又は自宅でのヘミン静脈内投与を要したポルフィリン症発作が2回以上認められた患者
- 肝機能障害患者[アラニンアミノトランスフェラーゼ(ALT)値がULNの2倍超、総ビリルビン(TBL)値がULNの1.5倍超、国際標準比(INR)が1.5超のいずれかに該当する患者]及び腎機能障害患者(eGFRが30mL/min/1.73m2未満の患者)を除外
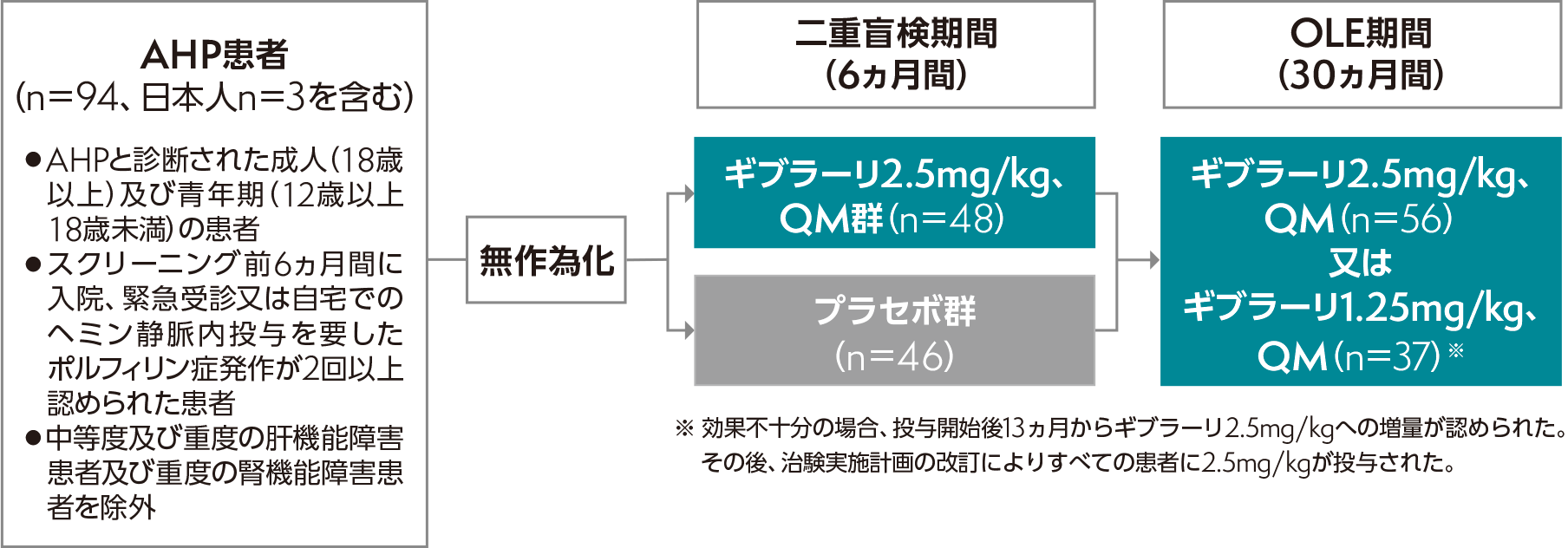
試験デザイン・投与方法
第Ⅲ相、多施設共同、無作為化、二重盲検、プラセボ対照、国際共同試験及びオープンラベル継続投与(OLE)試験
本試験は二重盲検期間及びOLE期間から構成される。
- 二重盲検期間
- ギブラーリ群又はプラセボ群に無作為に割り付け(1:1)、ギブラーリ2.5mg/kgもしくはプラセボを月に1回(QM)、6ヵ月間皮下投与した。
- OLE期間
- 二重盲検期間を完了した患者にギブラーリ1.25mg/kg又は2.5mg/kgをQMで30ヵ月間皮下投与する。なお、ギブラーリ1.25mg/kg投与患者において、効果が不十分であった場合には投与開始後13ヵ月(OLE期間で6ヵ月間のギブラーリ1.25mg/kg投与完了後)からギブラーリ2.5mg/kgへの増量を可とした。
試験期間を通して、急性発作に対するヘミン投与は可としたが、ヘミン予防投与は不可とした。
評価項目
主要評価項目
-
二重盲検期間におけるAIP患者のポルフィリン症複合発作*の年換算発作発現回数(AAR)
- *
-
入院(病棟への入院又は救急科受診で24時間以上の滞在に至った場合)、緊急受診(診療所、点滴センター又は救急科での緊急かつ予定外の受診であり、入院の基準を満たさないもの)又は自宅(入院又は緊急受診の基準を満たさない場所)でのヘミン静脈内投与を要した発作
国際共同第Ⅲ相臨床試験(ENVISION試験)では、ポルフィリン症発作を次の3つの基準をすべて満たすものと定義した。
- 腹部、背部、胸部又は四肢の内臓神経痛の急性発作
- 患者に通常日常的に行われているポルフィリン症管理以上の用量又は頻度で、デキストロースもしくはヘミンの静脈内投与、炭水化物、鎮痛薬(オピオイド[合成及び非合成物質]もしくは非オピオイド)又は制吐薬などの他の薬剤による治療を必要とする
- ポルフィリン症発作以外の他の原因を医学的に判断できない
副次評価項目
- 投与開始後3ヵ月、6ヵ月におけるAIP患者/AHP患者の尿中ALA値
- 投与開始後6ヵ月におけるAIP患者/AHP患者の尿中PBG値
- 二重盲検期間におけるAIP患者/AHP患者のヘミンの年換算投与回数
- 二重盲検期間におけるAHP患者のポルフィリン症複合発作のAAR
- 二重盲検期間におけるAIP患者/AHP患者の毎日の最悪疼痛スコア、毎日の最悪疲労スコア、毎日の最悪悪心スコア
- 投与開始後6ヵ月におけるAIP患者/AHP患者のSF-12(12-item short-form health survey)によるPCS(身体的側面のQOLサマリースコア)のベースラインからの変化量
探索的評価項目
二重盲検期間又はOLE期間におけるAIP患者/AHP患者の以下の評価項目
- ポルフィリン症複合発作の発現回数 など
安全性評価項目
- AHP患者の有害事象、臨床検査値 など
解析計画
解析対象集団
有効性評価項目の解析には最大の解析対象集団(FAS)及びAIP患者の最大の解析対象集団(FASAIP)を用い、二重盲検期間の安全性の解析には安全性解析対象集団を用いた。ギブラーリ投与期間中*の長期有効性及び安全性の解析には、全ギブラーリ投与解析対象集団を用いた。
- *
- 二重盲検期間又はOLE期間でのギブラーリ初回投与以降のギブラーリ投与期間
| FAS(full analysis set) | 無作為化され、治験薬を1回以上投与されたすべての患者 |
|---|---|
| FASAIP(AIP patients in full analysis set) | 無作為化され、治験薬を1回以上投与されたすべてのAIP患者 |
| 安全性解析対象集団 | 治験薬を1回以上投与されたすべての患者 |
| 全ギブラーリ投与解析対象集団 | ギブラーリを1回以上投与されたすべての患者 二重盲検期間にギブラーリを投与された患者と、二重盲検期間にプラセボを投与されOLE期間にギブラーリ投与に切替えた患者を含めた |
解析方法
二重盲検期間におけるAIP患者/AHP患者のポルフィリン症複合発作のAARは、投与群、層別因子(ヘミン予防投与歴及び過去の発作回数、AIP患者のみ)を固定効果、各患者の経過期間の対数をオフセット変数とする負の二項回帰モデルを用いて解析を行った。
二重盲検期間におけるAIP患者/AHP患者のヘミンの年換算投与回数は、投与群、層別因子(ヘミン予防投与歴及び過去の発作回数)を固定効果、各患者の経過期間の対数をオフセット変数とする負の二項回帰モデルを用いて解析を行った。
二重盲検期間におけるAIP患者/AHP患者の尿中ALA値及びPBG値は、ベースライン値を連続共変量とし、層別因子(ヘミン予防投与歴及び過去の発作回数)、来院、投与及び来院と投与の交互作用を固定効果、患者を変量効果とするmixed-effect model repeated measures(MMRM)を用いて解析を行った。
二重盲検期間におけるAIP患者/AHP患者の毎日の最悪疼痛スコア、毎日の最悪疲労スコア、毎日の最悪悪心スコアについては、週平均スコアのベースラインから6ヵ月間の変化量のarea under the curve(AUC)及びベースラインから6ヵ月間の週平均スコアの変化量を、投与群、層別因子(ヘミン予防投与歴及び過去の発作回数)を固定効果、ベースラインの週平均スコアを共変量とした共分散分析モデルを用いて解析を行った。
投与開始後6ヵ月におけるAIP患者/AHP患者のSF-12によるPCSのベースラインからの変化量は、ベースラインのスコアを連続共変量とし、投与群、層別因子(ヘミン予防投与歴及び過去の発作回数)、来院及び来院と投与の交互作用を固定効果とするMMRMを用いて解析を行った。
OLE期間における各評価項目の結果については、記述的に要約した。
試験全体での第一種の過誤確率を両側0.05に制御するため、非盲検下での中間解析の有意水準を両側0.001に調整して実施するとともに、主要評価項目及び副次評価項目の解析の有意水準を両側0.049として固定順序検定を実施した。予め設定された順序は、上記に記載の評価項目の順である。
有害事象は医薬品規制調和国際会議(ICH)国際医薬用語集(MedDRA)version 21.0を用いて器官別大分類及び基本語別に集計した。副作用は、治験責任医師が治験薬と「明らかに関連あり」「関連があるかもしれない」又は「おそらく関連なし」と判断した有害事象とした(治験薬との因果関係が評価されなかった有害事象も副作用に含まれる)。